Case presentation
A 39-year-old female was referred to ambulatory care with a one-week history of jaundice and nausea. She had been abstinent from alcohol for three months; however, previously she had been drinking six units of alcohol per day for five years. She had not travelled abroad recently. She was prescribed no regular medications but had been taking an over-the-counter herbal supplement containing basil powder, biotin and ashwagandha root extract on alternate days for the previous six weeks for self-management of anxiety. One capsule equated to 154 mg of ashwagandha root extract. On examination she was tachycardic and had jaundiced skin and sclera. Her abdomen was soft on palpation with no tenderness. She had no stigmata of chronic liver disease. Initial blood tests revealed a bilirubin of 154 µmol/L, alkaline phosphatase of 184 IU/L and alanine transaminase of 1,514 IU/L (Table 1). The R value was 26.7, confirming a hepatocellular pattern of liver injury. There were no abnormalities on full blood count or urea, and electrolyte testing and paracetamol level was undetectable. Further blood tests were sent for a non-invasive liver screen.
Table 1 Pertinent blood results from admission and follow-up.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Albumin
|
49
|
44
|
42
|
42
|
43
|
42
|
44
|
47
|
42
|
44
|
|
Bilirubin
|
8
|
154
|
220
|
252
|
279
|
293
|
327
|
292
|
182
|
115
|
|
Alkaline phosphatase
|
69
|
184
|
167
|
162
|
152
|
140
|
144
|
142
|
112
|
125
|
|
Alanine aminotransferase
|
14
|
1,514
|
1,494
|
1,503
|
1,443
|
1,316
|
1,301
|
995
|
520
|
495
|
|
Prothrombin time
|
n/a
|
14
|
13
|
n/a
|
14
|
13
|
14
|
12
|
12
|
12
|
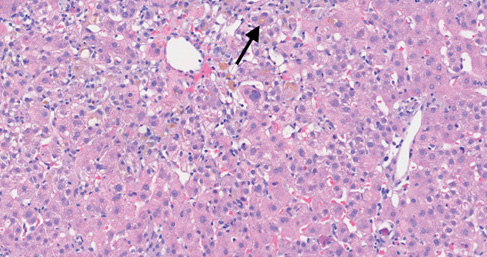
She subsequently developed right upper quadrant pain, lethargy and pruritus. An ultrasound scan revealed no acute abnormalities. In view of her increasing bilirubin level (Table 1), she was admitted to hospital. The initial working diagnosis included acute viral hepatitis, autoimmune hepatitis or DILI secondary to ashwagandha. The results of her blood tests revealed negative viral serology (Hepatitis A IgM, Hepatitis B surface antigen and core antibody, Hepatitis C antibody, Hepatitis E IgM, HIV antibodies and Covid-19 polymerase chain reaction). Anti-nuclear antibody titre was 1:40 and anti-mitochondrial, anti-smooth muscle and anti-liver kidney microsomal antibodies were negative. Immunoglobulins A, G and M, ferritin, caeruloplasmin and alpha fetoprotein were all within normal range. She was observed in hospital and monitored with daily blood tests. Her bilirubin continued to rise, peaking at 327 µmol/L, whereas her alkaline phosphatase and alanine transaminase had already peaked at initial presentation (Table 1). An ultrasound-guided liver biopsy revealed features of a severe acute cholestatic hepatitis with confluent necrosis but no convincing signs of chronicity (Figure 1). Although acute onset autoimmune hepatitis remained a possible diagnosis, it was felt that the most likely aetiology was ashwagandha DILI. In this case the Roussel Uclaf Causality Assessment Method score was 8, which implies that ashwagandha was a probable cause of DILI.
Figure 1 An acute cholestatic hepatitis, showing lobular disarray with a lymphocytic infiltrate and multiple foci of spotty necrosis. Bile pigment is seen in Kupffer cells and there is canalicular bilirubinostasis (arrow).
Different management options (observation, trial of steroids and trial of ursodeoxycholic acid [UDCA]) were discussed with the patient. She was advised to avoid ashwagandha-containing supplements in the future. A shared decision was made to start UDCA based on her weight (750 mg once daily). She was discharged from hospital with a plan to closely monitor her blood test results over the following weeks. Further clinical review and blood tests on an outpatient basis in the first two weeks after discharge revealed an ongoing but gradually reducing hyperbilirubinaemia and falling alanine transaminase (Table 1). Two weeks following discharge, she reported resolution of her previously reported symptoms.
Discussion
The Drug-Induced Liver Injury Network have reported a significant increase in the proportion of DILI cases attributable to herbal and dietary supplements.1 Ashwagandha (also known as Indian ginseng) is a herb extracted from the leaves or roots of Withania somnifera, a plant commonly found in India and Southeast Asia. It is often used in herbal supplements, and the active components, withanolides, are alleged to have neuroprotective, anti-inflammatory and anxiolytic properties. The concentration of withanolides present in the extract can vary, with extract from the leaves usually having higher concentrations than the root. Previous clinical trials have revealed no serious adverse effects, hepatotoxicity or elevation of serum enzymes related to ashwagandha.2 More recently, however, there have been several case reports that implicate ashwagandha in the aetiology of DILI, thus prompting LiverTox to place the herb in category C (probable cause of clinically apparent liver injury) for likelihood of DILI.3 Ashwagandha is often combined with other vitamins and herbs when used as a herbal supplement which can make it difficult to ascertain whether ashwagandha is responsible for possible DILI. In our case, ashwagandha was extracted from the plant root and combined with biotin and basil powder. Neither of these additional ingredients are known to have adverse effects nor are they linked to hepatotoxicity.
A recent case series describing five cases of DILI secondary to ashwagandha attempted to ascertain a clinical phenotype for this condition.4 Comparison of our patient’s clinical journey with those described in this case series revealed some similarities. The range of patients’ ages was 21–62 years old, with a mean age of 43 years old. All five patients in the case series developed jaundice between 2–12 weeks after starting ashwagandha supplementation, with some patients also reporting nausea, lethargy and pruritus. Liver injury was either cholestatic or mixed in nature and a prominent feature in each case was a prolonged period of hyperbilirubinaemia (5–20 weeks).
There were some notable differences identified between our patient and the cases previously described. Our case showed a hepatocellular pattern of liver injury in contrast to the mixed and cholestatic patterns described in the case series. Liver biopsy revealed features of a severe acute cholestatic hepatitis. There was significant confluent necrosis in zones 1 and 3 of the biopsy sample. The portal tracts were expanded and heavily inflamed, with a lymphohistiocytic infiltrate, and there was some interface hepatitis (Figure 1). In comparison, the only biopsy performed in the case series showed a purer cholestatic picture with prominent canalicular cholestasis and only mild mononuclear cell inflammatory infiltrate.4 The histopathological changes seen on biopsy reflect the hepatocellular pattern of injury identified in our case. The pattern of cholestatic hepatitis suggests that there is most likely an immune-mediated or idiosyncratic process rather than a direct hepatotoxic insult. It is not unusual for individual drugs to show heterogeneity in phenotype across cases of DILI. For example, co-amoxiclav can cause cholestatic, mixed and hepatocellular injury.5 It is possible that ashwagandha DILI similarly expresses heterogeneity in its phenotype.
Furthermore, our case is the first in the reported literature to have had hepatitis E excluded as a possible underlying aetiology. Hepatitis E infection can often have a very similar clinical picture to DILI and is an important condition to exclude when investigating acute liver injury.
Finally, we present only the second reported case of ashwagandha DILI where treatment with UDCA was trialled, and the first reported case where it was used as monotherapy. UDCA has multiple proposed mechanisms of action, including expanding the hydrophilic bile acid pool, increasing secretion of bile acids, and cytoprotection against the hepatotoxic effects of bile acids.6 UDCA has been utilised in the treatment of several cholestatic diseases, including primary biliary cholangitis and primary sclerosing cholangitis, although there is conflicting evidence regarding its benefit in all cases. Given that both biopsies reported from patients who developed ashwagandha DILI showed a cholestatic process, we felt it was reasonable to trial UDCA as treatment in this case, whilst acknowledging that high quality evidence to support treatment with UDCA in DILI is lacking.7 We saw rapid improvement in our patient’s hyperbilirubinaemia compared to the prolonged hyperbilirubinaemia reported in the case series. This may suggest that UDCA can be effectively used as a treatment option and may be an area of interest for further research.
Conclusion
This case adds important information to the limited literature surrounding ashwagandha DILI. The clinical features described in our patient have some similarities to previous case reports and hence provide further evidence for formulating a clinical phenotype for this condition. Our case also provides new information regarding the possibility of hepatocellular injury in patients with ashwagandha-induced liver injury. We have also demonstrated unique histological changes on liver biopsy. Additional cases with liver biopsy would be beneficial. Another avenue of interest would be the treatment of these patients with UDCA and whether this truly accelerates normalisation of hyperbilirubinaemia. Finally, our case report highlights the importance of considering herbal supplements as a possible aetiology when investigating patients with an acute liver injury whilst also ruling out more common pathologies such as viral hepatitis and autoimmune liver disease.