Introduction
Haemoglobinopathy is an inherited blood disorder caused by genetic mutation of the globin chains. It is characterised by reduced or absent synthesis of globin chains which leads to a spectrum of thalassaemia syndrome. It can also cause defects in the synthesis of the haemoglobin (Hb) protein structure, which gives rise to different types of Hb variants. To date, there is more than 1,000 Hb mutations that have been reported in the Globin Gene Server, Hb Var (http://globin.bx.psu.edu/hbvar).1 Certain types of genetic mutation in the Hb structure can alter its affinity for oxygen, which can lead to either a high oxygen affinity variant Hb or low oxygen affinity variant Hb. Patients with high oxygen affinity variant Hb can present with polycythaemia and thrombosis, whereas patients with low oxygen affinity variant Hb can present with cyanosis and low peripheral oxygen saturation.2 We report the first case of a family of three, diagnosed with low oxygen affinity variant Hb Cheverly in Malaysia.
Case presentation
Ms A, a 47-year-old female, presented to the hospital emergency department for sudden onset of palpitations whilst she was driving her car. It was associated with chest tightness and giddiness. She had no history of cough, reduced effort tolerance or syncope. She had hypertension and was on daily Amlodipine 5 mg tablets. She was otherwise physically fit and had never presented herself to the hospital.
At the hospital emergency department, her peripheral oxygen saturation (SpO2) was persistently at 81–85% under room air. She was otherwise comfortable and not tachypnoeic. Her blood pressure was 144/82 mmHg and pulse rate was 87 per min. Besides mild pallor, there was no cyanosis, jaundice, finger clubbing, lymphadenopathy and hepatosplenomegaly. In view of low peripheral oxygen saturation, she was given high-flow mask oxygen (HFMO2) 15 L/min. Her SpO2 did not improve, and she was referred to the anaesthesia department for impending ‘collapse’. She was subsequently transferred to tertiary hospital for further management. While waiting for her transfer, both her siblings (Ms B and Mr C) tested their oxygen saturation and were surprised to find both their readings were less than 85%, which they kept to themselves.
At our hospital, her pulse oximetry consistently showed readings below 85%. However, an arterial blood gas analysis showed good oxygenation with partial pressure of oxygen in arterial blood (PaO2) of 554.9 mmHg under HFMO2 15 L/min and PaO2 of 99.3 mmHg under room air. The arterial oxygen saturation (SaO2), methaemoglobin (metHb) and carboxyhaemoglobin (COHb) were normal. Full blood count (FBC) showed normochromic normocytic anaemia with haemoglobin of 10 g/dL. Peripheral blood smear and other haemolytic work up revealed no evidence of haemolysis. Her iron, vitamin B12 and folate levels were normal. She underwent a series of investigations to rule out lungs and heart pathology, which included troponin T, creatinine kinase-MB (CK-MB), 12 leads electrocardiogram (ECG), chest radiograph, transthoracic echocardiogram, 24-h Holter monitoring, computed tomography (CT) scan of thorax and pulmonary angiogram (CTPA), all of which showed normal findings. In view of diagnostic uncertainty, she was then referred to the haematology team for further evaluation. On the basis of these normal investigations, further investigations were carried out to rule out haemoglobinopathy, such as Hb variant, to explain for these pulse oximetry discrepancies.
Capillary electrophoresis was normal, with Hb A 96.7% and Hb A2 3.3%. High performance liquid chromatography (HPLC) showed Hb A 84.7%, Hb A2 3.3% and Hb F 0.5% (Table 1). There was a small peak at C window, with retention time of 5.16 min (Figure 1). Both her siblings were screened for haemoglobinopathy. Both were asymptomatic and had low peripheral oxygen saturation with SpO2 of 80–85% under room air. However, both had normal Hb and red cell count. HPLC of Mr C showed similar small peak at C window. DNA analysis for this family of three showed heterozygous mutation at codon 45 (TTT to TCT) of the beta globin genes via Sanger sequencing using big dye sequencing kit and analysed on the ABI 3730XL DNA analyser (Figure 2).
Table 1 Comparison of the pulse oximetry, full blood count, haemoglobin analysis and DNA analysis of the three siblings
| |
|
|
|
|
|
Pulse oximetry (%)
|
81–85
|
80–85
|
80–85
|
|
|
Full blood count
|
|
|
|
|
|
Haemoglobin (g/dL)
|
10.0
|
12.1
|
13.9
|
12.0–15.09 (female)
13.0–17.0 (male)
|
|
RBC (106/uL)
|
3.19
|
3.78
|
4.39
|
3.8–4.8 (female)
4.5–5.5 (male)
|
|
MCV (fL)
|
90.6
|
94.4
|
93.6
|
83.0–101.0
|
|
MCH (pg)
|
31.3
|
32.0
|
31.7
|
27.0–32.0
|
|
Peripheral blood smear
|
No haemolytic anaemia
|
No haemolytic anaemia
|
No haemolytic anaemia
|
|
|
HPLC
|
|
|
|
|
|
HbA (%)
|
84.7
|
83.4
|
85.3
|
|
|
HbA2 (%)
|
3.3
|
3.4
|
3.1
|
2.3–3.3
|
|
HbF (%)
|
0.5
|
0.4
|
0.3
|
<1
|
|
C window
|
Small peak at
5.16 min
|
No peak
|
Small peak at
5.15 min
|
|
|
Capillary electrophoresis
|
|
|
|
|
|
Hb A (%)
|
96.7
|
97
|
97.1
|
96.8–97.8
|
|
Hb A2 (%)
|
3.3
|
3.0
|
2.9
|
2.2–3.2
|
|
DNA analysis
|
|
|
|
|
|
Alpha globin gene
|
Normal
|
Normal
|
Normal
|
|
|
Beta globin gene
|
Heterozygous state of codon 45 (TTT>TCT)
Hb Cheverly is identified
|
Heterozygous state of codon 45 (TTT>TCT)
Hb Cheverly is identified
|
Heterozygous state of codon 45 (TTT>TCT)
Hb Cheverly is identified
|
|
| |
|
|
|
|
Figure 1 HPLC shows normal levels of Haemoglobin F and Haemoglobin A2 for Ms A (top) and Mr C (bottom). A small peak at C window (arrow) is observed for these two patients, with retention time of 5.16 min and 5.15 min, respectively.
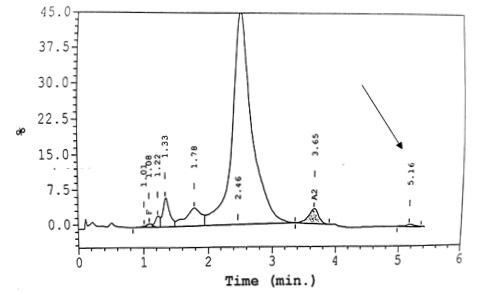
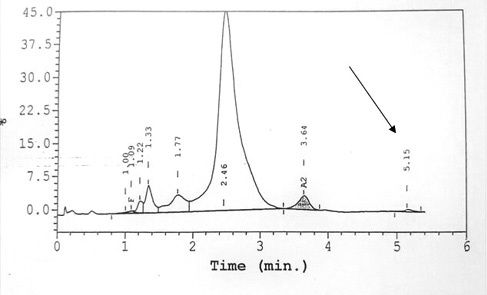
Figure 2 The snapshot of Sanger sequencing of beta globin gene reveals a point mutation at codon 45, with the substitution of thymidine to cytosine and, as a result, the replacement of amino acid phenylalanine by serine (Phe > Ser, TTT > TCT). The location of this point mutation in the beta globin gene cluster is at exon 2, which along with part of intron 1, is illustrated by the boxes above the sequence. The reference sequence is also shown at the top.
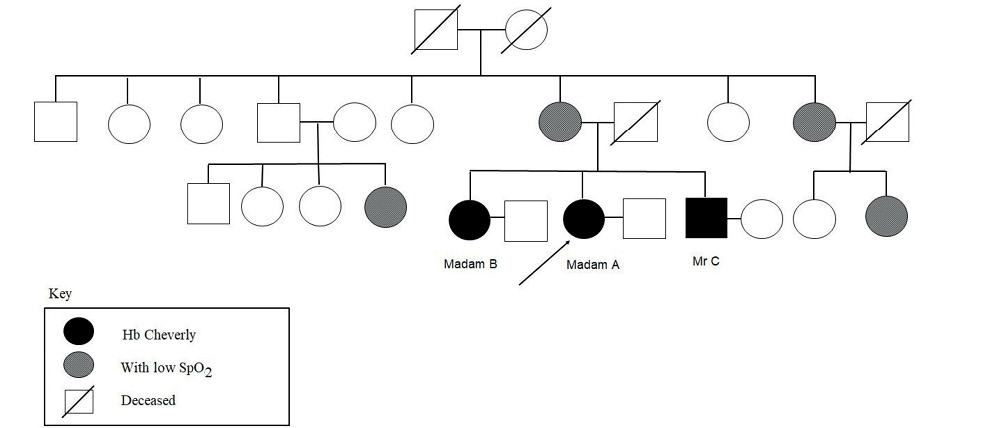
A diagnosis of low oxygen affinity variant Hb Cheverly was made. They were counselled and reassured for spuriously low peripheral oxygen saturation and advised for screening of the immediate and extended families. Only two of her cousins and one of her maternal aunts proceeded with pulse oximetry testing and had low SpO2 ranging from 76–85% under room air (Figure 3). However, they refused further blood taking.
Figure 3 Family tree of Ms A, Ms B and Mr C
Discussion
Haemoglobin protein is a tetramer that consists of two alpha and two beta globin chains. It is responsible in transporting oxygen from the lungs to peripheral tissues and facilitating the removal of carbon dioxide. When there is a structural defect in the synthesis of the Hb protein, it can compromise its ability to bind oxygen and result in low oxygen affinity Hb variant.3,4 In this case series, we describe a family of three that presented in Sarawak General Hospital, Malaysia, with low peripheral oxygen saturation and subsequently diagnosed to have Hb Cheverly. To date, there are few literatures that report patients with Hb Cheverly.
It was first described in 1982 in an elderly Italian male who had cyanosis since childhood and subsequently suffered from cardiomyopathy in his fifties. Another case in 1983 involved a family of three from Baltimore with chronic mild anaemia.5,6 An observational study done in Germany showed that only 9 out of 34,228 persons had Hb Cheverly.7 Hb Cheverly is caused by point mutation with the replacement of thymidine by cytosine and subsequently the substitution of the amino acid phenylalanine (Phe) by serine (Ser) at the CD4 region in the haem pocket, at codon 45 of the beta globin chain.6 This weakens the Hb interaction, rendering the instability of the mutated beta globin chain, leading to a mild haemolytic anaemia. Hb Hammersmith shares the similar amino acid substitution (Phe>Ser) but disrupts the CD1 region in the haem pocket, at codon 42 in the beta globin chain. However, Hb Hammersmith has a more severe Hb instability compared to Hb Cheverly, perhaps due to the dramatic alteration of the haem pocket which may affect the Bohr effect release of oxygen from the Hb.8,9
This case has highlighted the difficulty of diagnosing Hb Cheverly. It is discovered unintentionally during medical assessment for unrelated complaints. The clinical presentation of discordant SpO2 and PaO2 with mild normochromic normocytic anaemia give rise to the possibility of an Hb variant. We suspected Hb Constant Spring when the HPLC showed abnormal Hb at the C Window;10 however, DNA analysis of the alpha globin gene was normal and no mutation was detected. A diagnosis of Hb Cheverly was made based on the heterogenous mutation at codon 45 detected at the beta globin gene.
A pulse oximeter is commonly used to monitor oxygen saturation in our medical practice; however, it may not be an accurate tool to measure peripheral pulse oxygenation of patients with Hb variants such as Hb Cheverly. Hb Cheverly increases absorbance to between 600 nm and 660 nm, leading to an erroneous measurement of an increased proportion of deoxyhaemoglobin and subsequently spuriously low peripheral oxygen saturation.11 Early detection of such haemoglobinopathy is beneficial for the clinicians and the patients, and can avoid the unnecessary panic amongst clinicians in emergency settings. As for patient, it will spare them, and their affected family members, from undergoing unnecessary quarantine, diagnostic and therapeutic procedures. This scenario is further complicated in midst of the COVID-19 pandemic and ultimately causes distress to patients, family members and the medical team.
Conclusion
In summary, we need to consider Hb variants as part of our differential diagnosis in clinically well patients who present with unexplained low SpO2 after ruling out the more common cardiopulmonary causes. Good history recording, clinical examination and basic laboratory investigations can lead us to suspect an Hb variant which can later be confirmed by more advanced testing. This can avoid unnecessary panic and prevent unnecessary invasive diagnostic testing and intervention, especially in emergency settings. After establishing diagnosis, apart from issuing emergency cards, we have to counsel the patient and their caretakers and screen for possible affected family members.