Introduction
Cerebral salt wasting (CSW) is a syndrome that presents with hypovolaemic hyponatraemia and an elevated urine sodium in the setting of a disease of the central nervous system (CNS). CSW is most often described in patients with subarachnoid haemorrhage,1 but it has also been described in patients with CNS infections like meningitis, encephalitis, CNS tumours, surgery and head injury.2,3 The mechanisms by which CSW causes hyponatraemia are twofold: the first is impaired sympathetic neural input to the proximal tubule leading to reduced proximal sodium and urate reabsorption, and the second is an increase in circulating brain natriuretic peptide which further decreases sodium reabsorption and inhibits the release of renin.4
Catatonia is a behavioural syndrome seen usually in the context of psychiatric illnesses. Medical conditions related to catatonia include infectious diseases, electrolyte abnormalities, connective tissue diseases and neurological disorders.5 It usually presents as a motor abnormality in which a patient becomes incapable of normal motor activity despite having adequate power. Common features include immobility, abnormal posturing, waxy flexibility, purposeless movements or even marked agitation.
The case presented by us represents an unusual presentation of hyponatraemia caused by CSW leading to catatonia. Although there have been isolated reports of patients presenting with catatonia in the presence of hyponatraemia, most have been in the background of psychiatric illness or the hyponatraemia seems to have been triggered by an offending drug.6–9 We believe that this is the first instance of CSW-induced hyponatraemia presenting as catatonia.
Case presentation
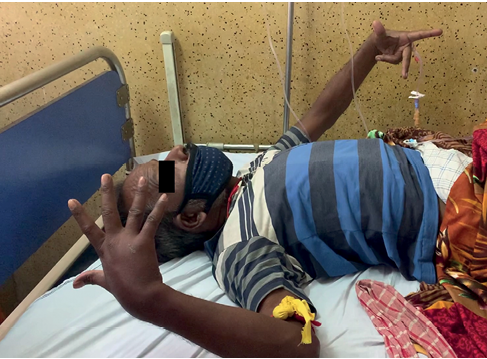
A 46-year-old male was brought to our Emergency Department with a five-day history of hiccups, followed by abnormal behaviour and posturing for one day prior to admission. He had been diagnosed with tuberculous meningitis 18 months ago and had been on anti-tubercular treatment (Rifampicin 450 mg, Isoniazid 225 mg, Pyrazinamide 1,200 mg and Ethambutol 825 mg for two months, followed by Rifampicin 450 mg, Isoniazid 225 mg, and Ethambutol 825 mg for ten months) until six months ago. During the first two months on anti-tubercular treatment, he had also received oral corticosteroids (Dexamethasone 24 mg/day for three weeks, 16 mg/day for two weeks, 8 mg/day for two weeks and 4 mg/day for one week). He had no other systemic illnesses, nor had he been on any medications for the last six months. On initial clinical examination we found the patient to be disoriented, immobile and mute with waxy flexibility and posturing (Figure 1) suggestive of catatonia (based on DSM-5 guidelines). He had poor skin turgor, sunken eyeballs and a dry tongue, with a pulse rate of 90 beats per minute, blood pressure of 112/86 mmHg and he was afebrile. There was no evidence of head injury, tongue bites, urinary incontinence or abnormal eye movements. Both pupils were equally reactive to light. His respiratory, cardiovascular and gastrointestinal system examination was unremarkable. Our initial suspicion included a reactivation of his meningitis, metabolic encephalopathies and stoke. In the setting of a pre-existing CNS disease, we opted to withhold a lorazepam challenge test to not risk further sedation or respiratory depression.
Figure 1 Patient at the time of presentation
His initial investigations (Table 1) revealed a plasma osmolality of 240 mOsm/L, serum sodium of 120 mEq/L, serum potassium of 3.5 mEq/L, urine osmolality of 184.46 mOsm/kg, a spot urine sodium of 95.5 mEq/L, a spot urine potassium of 22 mEq/L and a 24-hour urine volume of 6,000 mL. As he was dehydrated (confirmed by clinical response to isotonic saline fluid challenge test), a diagnosis of hypovolaemic hyponatraemia was made. Our differential diagnoses included salt wasting nephropathies, adrenal insufficiency, ketonuria, diuretic overuse and CSW. A baseline renal function revealed a blood urea of 43 mg/dL and a serum creatinine of 1.4 mg/dL, and urinalysis was normal. An abdominal ultrasound was obtained that showed structurally normal kidneys. Serum aldosterone at the time of admission was within normal limits at 15.7 ng/dL and urine ketone bodies were negative. The patient’s relatives denied the use of any medications in the last six months. Magnetic resonance imaging of the brain was performed, which showed mild hydrocephalus and basal-enhancing exudates (sequelae of tuberculous meningitis). In the setting of hypovolaemic hyponatraemia, structural CNS disease and corresponding blood and urine biochemistries, we considered CSW as the most likely diagnosis.
Table 1 Investigations at presentation
|
|
|
|
|
Haemoglobin (g/dL)
|
10.3
|
13.0–16.0
|
|
Total leucocyte count
(cells/mm3)
|
7,400
|
4,000–11,000
|
|
Haematocrit (%)
|
30.4
|
42–52
|
|
Aspartate transaminase (U/L)
|
78
|
5–40
|
|
Alanine transaminase (U/L)
|
49
|
5–40
|
|
Blood urea (mg/dL)
|
43
|
15–45
|
|
Serum creatinine (mg/dL)
|
1.4
|
0.9–1.3
|
|
Plasma osmolality (mOsm/L)
|
240
|
275–290
|
|
Serum sodium (mEq/L)
|
120
|
135–145
|
|
Serum potassium (mEq/L)
|
3.5
|
3.5–4.5
|
|
Serum calcium (mmol/L)
|
1.12
|
1.15–1.30
|
|
Urine osmolality
(mOsm/kg)
|
184.46
|
–
|
|
Urine sodium (mEq/L)
|
95.5
|
<20
|
|
Serum aldosterone
(ng/dL)
|
15.7
|
2.5–39.2
|
|
Plasma renin activity
(ng/mL/hr)
|
1.26
|
0.15–2.33
|
|
Serum uric acid (mg/dL)
|
4.0
|
4.0–8.5
|
| |
|
|
We commenced treatment with isotonic and hypertonic saline to improve volume status and serum sodium levels. Catatonia resolved on day 2 of admission, at serum sodium levels of 127 mEq/L. As serum sodium levels gradually improved, our patient had a dramatic improvement in mentation, and by day 4 of admission he became completely oriented and was able to follow verbal commands. He was discharged in stable condition on day 6 with a corrected serum sodium of 137 mEq/L. At a six-month follow-up, the patient continued to remain asymptomatic with no further episodes of hyponatraemia or catatonia.
Discussion
Hyponatraemia is defined as a serum sodium <135 mEq/L. Volume status of the patient plays a key role in determining the various aetiologies of hyponatraemia and, for this reason, hyponatraemia can be further classified as being hypervolaemic, euvolaemic or hypovolaemic. Hypotonicity caused by hyponatraemia produces cerebral oedema and this is responsible for neurological dysfunction in these patients. The most common causes of hyponatraemia include the syndrome of inappropriate antidiuretic hormone (SIADH), diuretic use and gastrointestinal losses (vomiting, diarrhoea).
Hyponatraemia-associated catatonia is a rare entity. It has previously been reported in the setting of psychiatric illness, drugs intake such as venlafaxine, imiquimod and ‘ecstasy’, and medical disorders like adrenal insufficiency.6–10 The mechanism by which hyponatraemia causes catatonia remains a matter of controversy. Novac et al. hypothesised that self-related processing is attributable to subcortical midline structures.7 These structures are interconnected with higher brain structures in the subcortical and cortical areas. It is likely that neurochemical dysfunction produced during hyponatraemia interferes with self-related processing (and the vertical processing system, of which it is a part), and is responsible for catatonia.
Interestingly, while some patients recover with correction of the hyponatraemia alone,11 others require different modalities of therapy, including benzodiazepines and electroconvulsive therapy.10 This highlights the importance of recognising the significant impact that electrolyte abnormalities may cause upon one’s mental status. Moreover, correction of the electrolyte abnormality may sometimes be inadequate to reverse the neuropsychiatric dysfunction.
In our patient, we attribute the hyponatraemia to CSW and he responded to salt and fluid correction. The existence of CSW has been a matter of debate amongst some authorities.12 It has often considered a form of the more common SIADH. Regardless, it is important to distinguish these two entities, as both have opposite treatment approaches. While SIADH requires fluid restriction, the goals of CSW treatment are volume and sodium replacement which can be done using isotonic saline, hypertonic saline and mineralocorticoids.13 The presence of volume depletion favours CSW rather than SIADH, in which the patient is likely to be euvolaemic. The possibility of CSW should be considered in all patients presenting with hypovolaemic hyponatraemia in the background of a neurological disorder. Although CSW has been reported to present with any of the numerous symptoms of hyponatraemia such as confusion, stupor, abnormal behaviour and seizures, the association of catatonia is yet to be reported. Keeping this in mind, clinicians should be vigil about its possibility in cases of CSW.
Conclusion
This case illustrates the importance of evaluating all patients with catatonia for electrolyte abnormalities, especially hyponatraemia. Furthermore, the possibility of CSW should be kept in mind in all cases presenting with hypovolaemic hyponatraemia when there is associated neurological disease. In such cases, timely correction of the electrolyte abnormality usually leads to excellent outcomes, precluding the need for benzodiazepines or electroconvulsive therapy.